“722”以来到底有多少品种撤回了!
- 2017-07-21 16:14
- 作者:CFDA
- 来源:国家食品药品监督管理总局食品药品审核查验中心网站
药物临床试验数据核查阶段性报告
(2015 年 7 月-2017 年 6 月)
为落实国务院《关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44 号)要求,国家食品药品监督管理总局(以下简称总局)于2015年7月发布了《关于开展药物临床试验数据自查核查工作的公告》(2015年第117号)。两年来,在总局的坚强领导下,国家食品药品监督管理总局食品药品审核查验中心(以下简称核查中心)认真履行检查职责,不断提升检查工作的规范性和统一性,共派出185个检查组对313个药品注册申请进行现场核查,圆满完成了总局确定的阶段性目标。
一、公告发布情况
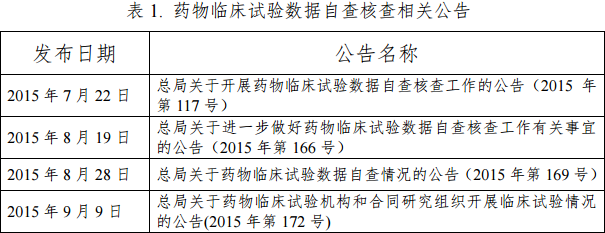
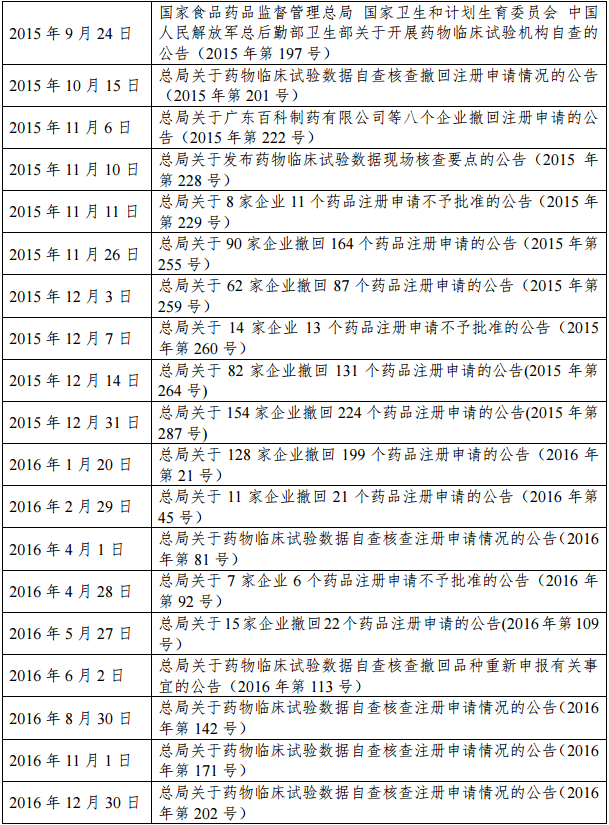
自2015年7月22日起,总局发布了27个公告,指导并通报临床试验数据核查工作的开展情况(见表1)。

二、品种填报及撤回情况
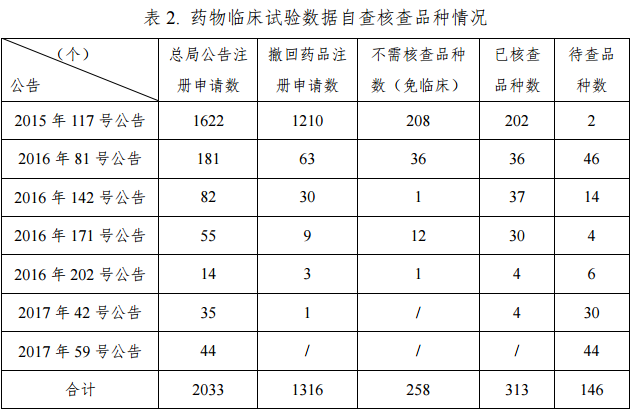
截止到2017年6月底,总局共发布 7 期公告,决定对2033个已申报生产或进口的待审药品注册申请开展药物临床试验数据核查。其中,申请人主动撤回的注册申请 1316 个,占 64.7%;申请减免临床试验等不需要核查的注册申请258个,占12.7%(见表2)。
三、核查工作进度
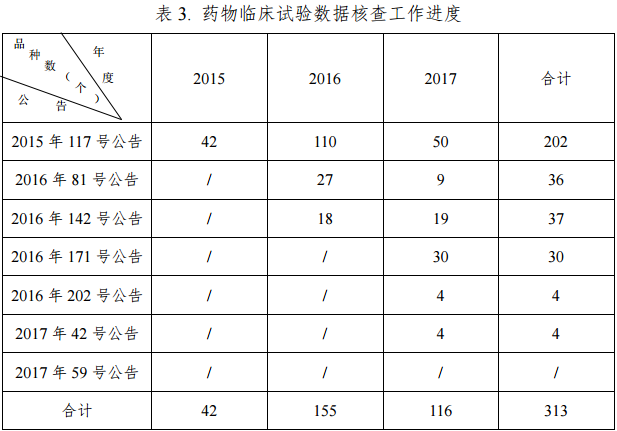
截止到2017年6月底,核查中心共派出检查组 185 个,组织检查员1635人次,对313个(见表 3)已提交自查资料的药品注册申请进行了临床试验数据现场核查,其中新药注册申请 94 个,仿制药注册申请 37 个,进口药注册申请 182 个。核查药物临床试验机构 763 家次,生物样本分析单位121家次,分布于全国28个省、自治区和直辖市。
四、核查结果处理情况
在已核查的313个药品注册申请中,有38个注册申请的临床试验数据涉嫌数据造假,其中新药注册申请 16 个,仿制药注年度品种数(个公)告5册申请17个,进口药注册申请5个。总局已发布公告,对其中30个注册申请作出不予批准的决定,并对其中涉嫌数据造假的11个临床试验机构及合同研究组织(CRO)予以立案调查。其余8个注册申请的核查资料正在按程序处理。
五、核查工作程序
药物临床试验数据核查工作在核查中心质量管理体系下进行,在检查员培训、检查员利益冲突管理、现场检查准备、现场检查实施、检查结果报告、检查资料存档等环节都制定有工作程序,并对现场核查过程进行质量监控。
(一)制定现场核查计划
药审中心根据审评进度和评价需要,向核查中心提供需要核查的品种情况。核查中心按审评顺序、自查报告填报情况以及举报信息等情况拟定现场核查计划。
(二)核查计划公示
从 2016 年 3 月至 2017 年 6 月底,共发布药物临床试验数据现场核查计划公示 12 期 351 个品种,公示期为 10 个工作日,在公示后 10 个工作日内未提出撤回申请的视为接受现场核查。
(三)开展现场核查
检查组根据《药物临床试验数据现场核查要点》(2015 年第228 号公告)进行核查,确保各组间尺度一致。根据注册申请类6型,II/III 期临床试验平均检查试验场点 4 个,平均检查时间 7 天;生物等效性试验检查临床部分与生物样本分析部分,平均检查天数 4 天。
(四)集中会审
核查中心建立了由临床医学、药学、生物分析、医学检验、医学统计、医学伦理等各专业专家组成的委员会,采用盲审的方式对核查结果进行集中会审,确保会审结果客观公正。
(五)反馈沟通核查情况
集中会审后,核查中心向药品注册申请人和主要研究者反馈和沟通核查情况。如有异议可进一步提交资料,申请二次会审。截至 2017 年 6 月底,共组织召开沟通会 14 次,参会人员超过 920 人次,答疑近 500 条。
(六)核查意见转药审中心
综合核查意见与沟通情况,核查中心形成明确的核查意见,随相关材料转药审中心进行综合审评。现场核查开始到核查意见转药审中心的平均时间为 24 个工作日。
六、发现主要问题
(一)现场核查发现缺陷的总体情况
经对 313 个药品注册申请的现场核查报告进行分析,共发现5111 条缺陷项。其中临床部分 4583 条,平均每个临床试验机构7发现问题 6 条;生物分析部分 528 条,平均每个生物样本分析单位发现问题 4.4 条。依据《药物临床试验数据现场核查要点》对缺陷进行分类,发现缺陷条款数量最多的部分依次为:临床试验过程记录及临床检查、化验等数据溯源方面(占 28.1%)、方案违背方面(占 12.0%)、试验用药品管理过程与记录方面(占 11.6%)和安全性记录、报告方面(占 10.1%),共发现缺陷 3161 项,占61.8%(见图 1)。
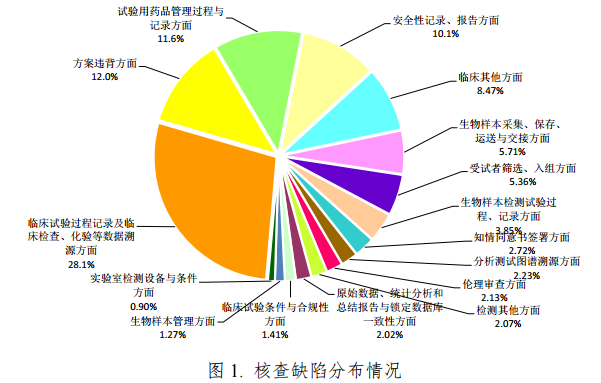
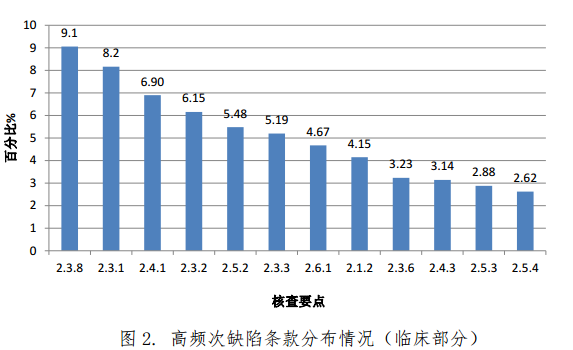
(二)高频次缺陷条款分布情况
对临床部分现场核查发现的 4583 项缺陷汇总分析,频次最高的缺陷条款见图 2。
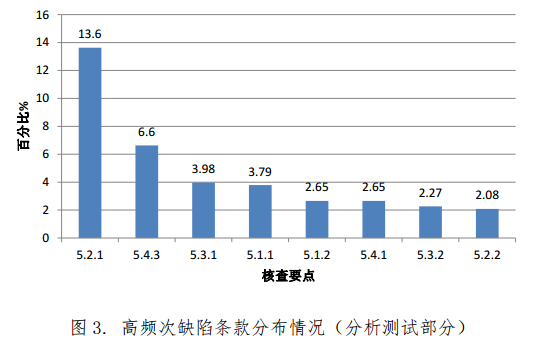
对分析测试部分现场核查发现的 528 项缺陷汇总分析,频次最高的缺陷条款见图 3。
(三)发现主要问题
临床试验过程记录及临床检查、化验等数据溯源方面
受试者体检单上性别栏显示为男性,但 X 线片图像显示为女性;报告单总的报告时间和人员信息被裁掉;脉搏治疗前后一致,不合常理;研究病历显示受试者已死亡,仍继续有访视记录和检验报告;多例受试者试验的血生化化验单中的部分数据(主要为肝肾功能指标)与医院 LIS 系统中的数据存在严重差异;
临床试验仅有病例报告表,无原始病历记录;
数据不可溯源,受试者血常规、尿常规等数据无法溯源;
不同受试者入出组的血生化报告值完全相同;
心电图图谱、报告真实性存疑,同一受试者试验前后波形不一致;
主要疗效指标、次要疗效指标无法溯源等等。
中心实验室样品放置时间超过样品在该条件下的稳定时间;样品运输过程中无温控记录;检测报告单上无检测人和复合人的签名;样品报告时间与样品接收间隔数天;样品被连续转包导致检测周期过长等。
2. 方案违背方面
修改入组检查结果,使其符合方案规定入组标准;
漏记方案规定禁用药物、合并用药;
给药方式、给药剂量及采血时间偏离试验方案;
10未按方案随访至规定日期等。
3. 试验用药品管理过程与记录方面
试验用药品不真实,参比制剂的包装规格和药片外观与试验制剂完全一致;
直接将试验制剂用作参比制剂,造成两者等效的假象;
药品发放和回收数量不一致;
药品管理员未被授权;
药品运输过程中的温度超出规定温度;
无药品回收记录;
药品接收和发放记录单无药品批号或批号不一致;
药品发放记录单中修改不规范等。
4. 安全性记录、报告方面
发生 AE,CRF 和总结报告中未录入;
修改 AE 与试验药物相关性的判断;
未按规定上报 SAE;
AE 漏记等。
5. 生物样本检测试验过程、记录方面
仪器还未购买就有了检测数据;缺乏生物样本预处理、保存、转运以及 LC-MS/MS 、离心机使等关键部分的记录;
样本分析过程记录原始记录缺失,相关记录为事后整理补充11填写,分析过程记录等是后期整理得到,没有原始分析记录;
隐瞒弃用试验数据,原始记录本中未记录;
图谱文件与血样没有关联性,两者之间的联系可随意更改且无法追溯;
部分报告数据与原始图谱计算所得数据不一致;
存在不合理手动积分等。
6. 分析测试图谱溯源方面
方法学验证及生物样本测试的稽查轨迹中均多处出现分析测试系统日期反复更改、重复检测后用同一文件名命名并覆盖原有图谱;
从质谱图谱中发现选择性使用符合要求的标准曲线和质控样品,并在 Audit trail 中发现将被采用的样本的序号( SampleID )修改成连续序列;
多个时间点样品编号与对应的图谱中的文件名编码从小到大的顺序颠倒;
部分受试者分析序列采集过程有中断,质控样本重复进样,部分样本重分析而没有随行质控,但数据被采用;
多个样本测试数据文件的采集时间有重叠;
仪器被转卖,分析测试图谱无法溯源;
选择性使用数据;
12分析测试图谱无法溯源等。
7. 伦理委员会主要问题
伦理委员会未对上报的 SAE 进行审查;
试验方案修改未经伦理委员会审查;
项目接收组长单位伦理批件,而未见任何相关的记录;
未见针对该项目伦理委员会会议审查的原始记录;
伦理委员会会议审查记录原始记录本内容与整理后的会议纪要内容不符等。
七、检查员培训
核查中心建立了检查员库,检查员均具有临床试验相关的资质和经验,专业背景涵盖临床医学、生物统计、生物分析、公共卫生等各专业,并经过系统性培训和考核。目前共有500 余名检查员参加了现场核查工作,检查员来自于核查中心、中检院、药审中心、药典委、各省局、各大专院校及医疗机构。核查中心制定有外部检查员选择和使用的管理办法,对保密性、独立性和利益冲突等进行管理。
除基础培训外,核查中心积极组织各种国内外培训,不断提高检查员整体素质水平。在每次检查前对检查组进行集中培训,统一标准和认识。
核查中心重视检查员队伍建设,加大招聘工作力度,将一大13批具有专业背景和研究经验的人员充实到检查一线岗位,制定有新检查员培训计划,努力建设职业化、专业化的检查员队伍。
八、总结与展望
两年来,在总局领导下,核查中心不断总结检查经验,创新检查手段,数据核查工作取得了明显成效。但是,也要清醒地看到,药物临床试验领域还存在不少问题,造假现象依然存在,数据核查工作任重而道远。
下一步,核查中心将按照总局要求,全面提升核查工作质量,重点做好以下工作:
(一)保持高压态势不松劲,继续做好数据核查工作
进一步完善现场核查技术标准和判定原则,加强与药审中心的沟通衔接,不断提高检查水平,确保尺度不放松,标准不降低。
(二)加强检查员能力建设
进一步充实检查员队伍,继续加强检查员的培训工作,选拔和培训检查组长和检查骨干,提高检查员的专业性、技术性、权威性。
(三)加强信息公开
加强核查信息公开,实现全过程的进度查询。扩展信息公开的内容和范围,提高信息公开的质量,接受社会监督。
(四)不断完善临床试验核查制度
14完善核查工作数据库,建立临床试验机构、申办者检查档案信息,探索基于风险的核查制度,提高核查效率。针对核查中发现的问题,研究建立对伦理委员会、申办者等的延伸检查机制。
临床试验数据是药品审评审批的依据,其真实性决定药品的安全性、有效性,直接关系公众用药安全。数据核查工作的开展,使临床试验造假的态势得到了明显遏制,净化了药物研发生态环境。在总局领导下,核查中心将继续以维护公众健康为己任,不忘初心、继续前进,做好药品安全守望者。
《中国医药报》社版权所有,未经许可不得转载使用。
(责任编辑:)
分享至
右键点击另存二维码!

-
相关阅读
-
为你推荐
-
第26期“安安有约——食药科普大讲堂” 美丽520:破解化妆品谣言正当时
5月20日,坊间颇具浪漫色彩的一天,广东省药品监督管理局主办的第26期“安安有约——食药科普大讲堂活动”在广州举行,化妆品专家蒋丽刚《破解化妆品谣言》的专题讲座也如期开讲。 2019-05-27 09:55 -
42a0b751-7d56-4b1c-a143-a99f7da2fa3c_260x150c.jpg)
科普大讲堂
2019-04-28 15:07








互联网新闻信息服务许可证10120170033
网络出版服务许可证(京)字082号
©京公网安备 11010802023089号 京ICP备17013160号-1
《中国医药报》社有限公司 中国食品药品网版权所有